MetaboHunter is a Python module for accessing the MetaboHunter web service web service for automated assignment of 1D raw, bucketed or peak picked NMR spectra.
More information about the algorithm is available in the published paper:
Tulpan, D., Leger, S., Belliveau, L., Culf, A., Cuperlovic-Culf, M. (2011). MetaboHunter: semi-automatic identification of 1H-NMR metabolite spectra in complex mixtures. BMC Bioinformatics 2011, 12:400
I find the service useful to give a first-pass identification of metabolites from 1D spectra, which can subsequently be confirmed or combined with identification via other methods. I originally wrote a Python interface as a standalone script, then as a Pathomx plugin, and have now moved the code into a reusable Python module with some extra IPython goodness. The walkthrough below demonstrates using the service with standard settings, passing a numpy array of ppms and peak heights. There is also a demo of a simple IPy Notebook widget set that can be used to configure the request.
The module and source code is available via PyPi and Github.
You can download a IPython Notebook to try it yourself.
Setup
The module is on PyPi and has no funky
dependencies. You should be able to install the metabohunter package
from the command line:
pip install metabohunter
To use the module simply import it. The main module object provides two
useful things: a request function that performs the request to the
MetaboHunter service and a IPyMetaboHunter which provides nice
widgets for IPython Notebooks and a synchronized config dictionary that
can be passed to requests.
import metabohunter as mh
import numpy as np
import os
os.environ['http_proxy'] = ''
Input format
To make a request to the MetaboHunter service you need to provide two lists (or 1D numpy arrays) of ppm values (the x axis scale on an NMR spectra) and peak heights (y axis). Here we create some dummy data using an 50-element axis of 0-10 in 0.2 increments, together with a 50-element series of peak heights generated randomly.
ppms = np.arange(0,10,0.2)
peaks = np.random.random(50)*10
The resulting output in ppms is:
array([ 0. , 0.2, 0.4, 0.6, 0.8, 1. , 1.2, 1.4, 1.6, 1.8, 2. ,
2.2, 2.4, 2.6, 2.8, 3. , 3.2, 3.4, 3.6, 3.8, 4. , 4.2,
4.4, 4.6, 4.8, 5. , 5.2, 5.4, 5.6, 5.8, 6. , 6.2, 6.4,
6.6, 6.8, 7. , 7.2, 7.4, 7.6, 7.8, 8. , 8.2, 8.4, 8.6,
8.8, 9. , 9.2, 9.4, 9.6, 9.8])
...and in peaks:
array([ 8.31680605, 6.04419835, 6.89353176, 6.00962915, 4.41208152,
3.2333172 , 1.39946687, 6.4614129 , 6.20912024, 0.06888817,
7.42894489, 6.7128017 , 0.79111548, 8.85208481, 4.9710428 ,
4.95762437, 9.82106628, 3.3606115 , 8.71282185, 9.6313281 ,
5.1396787 , 6.90228616, 4.12455523, 3.71683751, 1.77995641,
1.87159547, 5.43813402, 6.26325801, 9.17281811, 2.507874 ,
0.64188688, 5.03782693, 6.93223808, 8.59120112, 2.95107901,
9.70824585, 1.30386675, 1.02667654, 2.46923911, 9.02715511,
2.42110673, 5.2022395 , 8.79650171, 7.06068795, 9.45386543,
4.38466017, 0.22570328, 3.25368676, 0.63608104, 6.98335382])
Performing a request
The results are returned back in a list of the same length as the input
array. Mapped metabolites are represented by their Human Metabolome
Database (HMDB) identifier whereas unmapped peaks
are represented by None.
hmdbs = mh.request(ppms,peaks)
In hmdbs is now a list of metabolites using HMDB identifiers.
[None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
'HMDB00766',
None,
'HMDB00210',
'HMDB01919',
'HMDB01919',
None,
None,
'HMDB00210',
None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
'HMDB00763',
'HMDB00617',
'HMDB00763',
'HMDB00259',
None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
None,
None]
To throw away the None's and get the ppm values for the mapped metabolites you can do something like:
[(ppm, hmdb) for ppm, hmdb in zip(ppms, hmdbs) if hmdb is not None]
Which will give:
[(2.0, 'HMDB00766'),
(2.4000000000000004, 'HMDB00210'),
(2.6000000000000001, 'HMDB01919'),
(2.8000000000000003, 'HMDB01919'),
(3.4000000000000004, 'HMDB00210'),
(6.8000000000000007, 'HMDB00763'),
(7.0, 'HMDB00617'),
(7.2000000000000002, 'HMDB00763'),
(7.4000000000000004, 'HMDB00259')]
IPython Candy
To make the metabohunter module a bit nicer to work with from within
IPython Notebooks, the module provides a simple class for generating
widgets to control settings. The class is initialised with the default
settings for the request, however you can pass additional variables (any
of the keyword arguments allowed for request).
mhi = mh.IPyMetaboHunter(confidence=0.1, tolerance=0.5)
Once the object is created you can call .display() to render the
widgets in the current cell. Any changes to the variables are stored
back into the IPyMetaboHunter class object (here mhi) and
available in subsequent calculations.
mhi.display()
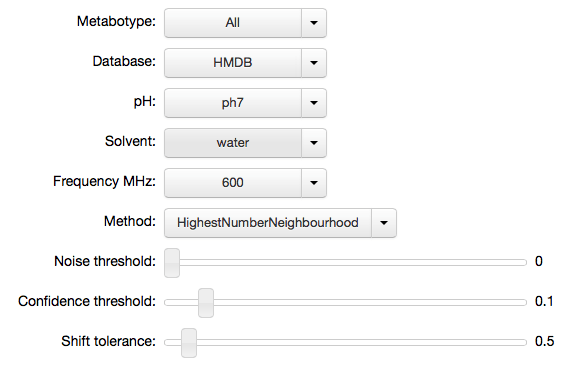
mhi.settings
{'confidence': 0.1,
'database': 'HMDB',
'frequency': '600',
'metabotype': 'All',
'method': 'HighestNumberNeighbourhood',
'noise': 0.0,
'ph': 'ph7',
'solvent': 'water',
'tolerance': 0.5}
The widgets manager makes the keyword arguments for the request
available via a kwargs property. To provide these to the request
function as keyword arguments we just need to unfurl it into the
function call using **. Try adjusting the parameters above and
seeing how they affect the results when re-running the request.
mh.request(ppms,peaks,**mhi.kwargs)
[None,
None,
None,
None,
None,
'HMDB00172',
'HMDB00011',
'HMDB00518',
'HMDB00510',
'HMDB00510',
'HMDB00518',
'HMDB00510',
'HMDB01547',
'HMDB01547',
'HMDB00101',
'HMDB00208',
'HMDB00192',
'HMDB00162',
'HMDB00014',
'HMDB00122',
'HMDB01401',
'HMDB00272',
'HMDB00902',
'HMDB00085',
None,
None,
'HMDB00215',
None,
'HMDB00393',
None,
None,
None,
None,
None,
'HMDB01392',
'HMDB00617',
'HMDB00303',
'HMDB01406',
None,
None,
'HMDB00232',
'HMDB00902',
None,
None,
None,
None,
None,
None,
None,
None]
Wrapping up
Hope you find this useful. If you have requests for features or improvements, feel free to get in touch. I'll be adding a walkthrough to the IPython widget-generator class in a future post.